
NCS - TBL - AULA 2 - 26/08/24
- thikow

- 26 de ago. de 2024
- 8 min de leitura
Atualizado: 21 de set. de 2024
TBL 2 - Distúrbios da proliferação e crescimento celular
Bloqueia Bloqueia
BH3 → BCL2-XL → BAX/BAK → Induzem a apoptose → Apoptose
Pró-apop Anti-apopt. Pró-apoptótica
Vias de ativação:
Via intrínseca (Mitocondrial) → Dano celular → P53 → BAX/BAK → ATC (citocr. C) → Ativa Caspases
Via extrínseca (Receptor da morte) → Rec. Morte (TNF) → Complexo da morte → Ativa Caspases
Quem ativa o TNF é o CD8 ou NK
BH3 é pró-apoptótico pois bloqueia o freio anti-apoptótico, logo, favorece a apoptose
Via extrínseca ativada quando LTCD8 e NK se liga ao receptor CD95 → Faz (Receptor expresso na maioria das células)

Uma parte da via intrínseca vai ser ativada pela via extrínseca via:
Caspase 8 ativa BID (BH3) → BH3 que bloqueia o BCL
BID é uma BH3 → Tem um domínio igual a BCL, logo, consegue bloquear a BCL.
Intrínseca → Ativada quando o dano a DNA gera um dano ao P53 ativando os genes BAX/BAK produzindo proteína BAX/BAK. Dentro da mitocôndria tem citocromo c. BAX/BAK altera a permeabilidade da membrana da mitocôndria, fazendo com que o citocromo c saia para fora. Ele no citoplasma forma um complexo com a proteína APAF-1 (proteína ativadora pró-apoptótica), ativar a Caspase 9, que por sua vez, ativa a Caspase 3.
Citocromo C = COX (Cicloxigenase oxidase)
Câncer tem superexpressão de BCL, logo, bloqueia BAX/BAK.
FAS (CD95) – Protéina quimérica que ativa a via de morte celular
A neoplasia ainda pode induzir a formação de uma proteína chamada c-FLIP → c-FLIP interage ao complexo sinalizador induzido por morte e não deixa ele ser ativado.
Se não tiver APAF-1 não forma complexo e não ativa Caspase-9
*Qual o problema do linfoma que superexpressa BCL2?
A superexpressão de BCL vai promover o bloqueio da apoptose através do bloqueio BAX/BAK
*Qual o problema de tumores que tem o TP53 mutado?
TP53 está relacionado com a ativação da via intrínseca que é responsável por ativar BAX/BAK que ativa a permeabilidade da membrana, não liberando citocromo c, logo, não formando complexo do citocromo c com APAF-1, não ativando caspase responsáveis pela morte celular.
*Qual a vantagem de pesquisar drogas que simula BH3 only?
BID bloqueia BCL2, logo consegue ativar BAX/BAK.
*Um pct fez uma testagem molecular. Tinha mutação em gene BID, como isso pode ativar a mutação da via intrínseca?
Pois BID bloqueia BCL2 que é anti-apoptótico, se tem mutação em BID não consegue bloquear BCL2, logo, a célula não consegue fazer a morte celular.
Célula na autofagia pode degradar componentes citoplasmáticos, lesando a mitocôndria e tirando problema. Porém quando a autofagia lesa um componente gera energia. Em estágios avançados do câncer ele pode estimular a autofagia para gerar energia para ele.
A autofagia pode ser induzida pelo câncer, principalmente durante a terapia com medicamentos, para gerar energia para ele.
Conforme a célula vai se dividindo os telômeros vão reduzindo. Nem toda a célula tem telomerase ativa para aumentar os telômeros.
Toda vez que tem encurtamento de telômeros vai dar problema → Precisa ativar p53 para jogar a célula para a senescência (pois o telômero muito encurtado não tem como voltar a se duplicar). Se os pontos de controle estiverem desabilitados o DNA vai fazer troca de bases através de novas quebras na dupla de DNA, gerando uma instabilidade mitótica.
Ausência de p53 faz que uma enzima uma as duas extremidades dos cromossomos, o que causa mutação e gera uma catástrofe mitótica. O câncer ativa a telomerase, para aumentar a ponta dos telômeros evitando a catástrofe mitótica.
A ativação da telomerase é observada praticamente em todos os cânceres.
Sem o p53, a célula vai fazer a ativação de salvamento e da via não homóloga de união das pontas (unindo as pontas dos cromossomos). Na anáfase quando for duplicar vai haver quebras na fita dupla levando a catástrofe mitótica (quando não tem telomerase) ou ao câncer (quando tem telomerase).
A superexpressão de telomerase é apresentada em um câncer mais agressivo, pois é mais rápido.
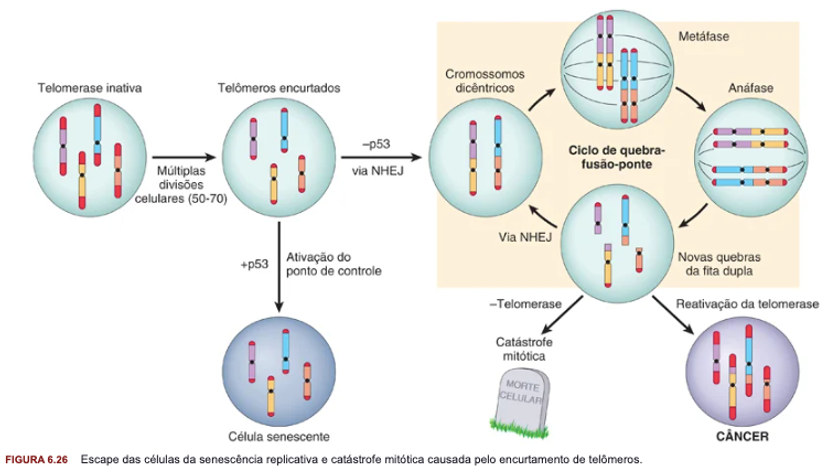
*Para a célula neoplásica conseguir ativar replicação ilimitada o que é necessário?
A célula tumoral muda o gene TP53 para evadir apoptose, quiescência e senescência
Tumores malignos crescem muito rapidamente, logo sua irrigação não será suficiente precisando estimular a angiogênese da região através de determinados mecanismos.
Para haver o crescimento de vasos precisa de fatores angiogênicos e reduzir/perder os inibidores da angiogênese. Fator de crescimento endotelial vascular (VEGF) é induzido pelo fator 1-alpha induzido por hipóxia (HIF-1alpha).
Tumor cresce → Hipóxia → HI1-1alpha → VEGF → Angiogênese
Normóxia → VHL se liga e degrada HIF1-alpha (feedback negativo)
Fatores pró-angiogênicos → VEGF (Estimulado pela hipoxia/HIF-1alpha)
Fatores inibidores → Trombospondina-1 (TSP-1), que é induzida pela p53
VHL faz feedback negativo em HIF-alpha através da degradação dele, logo, pode agir como supressor de tumor (não chega suprimento ao tumor).
p53 mutado → Celular perde apoptose, perde senescência, perde quiescência e perde controle de angiogênese.
Essa neoformação é de um vaso mais dilatado e com uma maior permeabilidade, logo é um vaso mais frágil que o original.
*Como VHL age como supressor de tumor
Degradando HIF-1alpha, logo desfavorecendo a angiogênese e a nutrição do tumor.
Capacidade de invadir e metastaziar
Células estão presas umas as outras através da E-caderina e ancoradas na MEC através de proteínas (colágeno tipo IV). Quando o colágeno IV é quebrado pelas proteases, a célula continua apresentando receptor para colágeno, logo, ela vai sendo atraída por ele e vai se locomovendo (é atraída por outros pontos) até chegar para dentro do vaso.
Uma das justificativas para metástase é perder a E-caderina.
A degradação das proteínas da MEC (de membrana basal) e do tecido conjuntivo intersticial é mediada por protease.
A clivagem (quebra) das proteínas da membrana basal, colágeno tipo 4 e laminina, por metaloproteinases estimulam a migração. Produz citocinas que vão deixar o receptor supersensível (aumentam a afinidade).
A migração parece ser potencializada e direcionada por citocinas derivadas das células tumorais, como os fatores de motilidade autócrina.
Resumo → Célula fixada nas proteínas de MEC e umas as outroas por E-caderina. Quebrando E-caderina aumenta a junção celular, tal como, proteases degradam as proteínas de ligação formando novos sítios de ligação. Migração são potencializados por fatores de motilidade autócrina (citocinas).

Nem toda metástase vai fazer o trajeto mais óbvio. Algumas tomam outros rumos atraídas por quimiocinas produzidas pelo corpo.
1- Afrouxamento das células através da quebra da E-caderina pela B-catenina. Célula produz metaloproteinases (proteases), que vão degradar as proteínas da MEC gerando novos pontos de ligação (sítios de ligação). Os produtos da clivagem do colágeno e proteoglicanos estimulam efeitos quimiotáticos, angiogênicos e promotores de crescimento. A clivagem de proteínas de membrana (colágeno IV e laminina) gera novos sítios que se ligam a receptores nas célilas tumorais e estimulam sua migração. O fator de motilidade autócrina facilita a locomoção da célula até chegar a corrente sanguínea. Continua produzindo protease, vai romper a parede do vaso e atravessar para dentro da corrente sanguínea. Até esse momento a célula esta suscetível a imunidade inata, porém pode se revestir de plaquetas para evadir a resposta imune.
Ø O tropismo do órgão pode ser relacionado a expressão das moléculas de adesão pelas células tumorais, cujos ligantes expressam de preferência no endotélio de órgão-alvos.
Ø Também é justificada pela célula cancerígena ter receptores para determinada quimiocinas presentes em determinadas regiões.
Ø Capacidade de colonizar o local Alguns ambientes podem ser desfavoráveis ao crescimento de semeaduras tumorais.
Porém não é possível precisar aonde a célula vai metastatizar, apenas prever e tentar rastrear.
Genes TWIST e SNAIL vão agir desregulando as E-caderinas
Reprogramação do metabolismo de energia
Célula desvia glicose a ácido lático mesmo na presença de oxigênio diferentemente das células normais do organismo para ter o crescimento mais rápido. Célula tumoral faz esse desvio, que geralmente ocorre só na falta de oxigênio, mesmo com o2 para obtenção de energia mais rápido.
Instabilidade genômica
Genes de reparo agem como corretores ortográficos trocando bases, evitando assim o pareamento errôneo das bases. São ativados pelo p53. Indivíduos com mutações nos genes de reparo tem um risco maior a desenvolver câncer. Sem ação dos genes de reparo os erros se acumulam a uma velocidade maior.
Embora os seres sejam expostos a agentes ambientais mutagênicos os cânceres são resultados raros desses encontros devido a ação de genes de reparo do DNA. Mutações em dois genes, BRCA1 e BRCA2 respondem por 50% dos casos de câncer de mama familiar.
Inflamação promotora de malignidade
Células de defesa produzem radicais livres para matar a bactéria. Produz espécie reativas de oxigênio (EROS) que liberados por macrófagos podem levar dano adicional a célula e favorecer dano adicional ao DNA.
Logo, células inflamatórias podem exercer atividades promotoras de tumor por produzir fatores de crescimento e infligir dano adicional ao DNA. A inflamação estimula proliferação de células e freia a apoptose. A persistente replicação e reduzida apoptose em um processo inflamatório tumoral aumenta o risco de adquirir mutações em um ou mais genes envolvidos nas carcinogêneses.
A expressão de cox2 é alta em cânceres de cólon e outros tumores, o que justifica pesquisas com aines no câncer.
Tumor com aumento de COX2 → Bloqueio de COX2 → Reduz inflamação → Reduz fator de crescimento e proliferação celular. Também vai reduzir EROS, diminuindo o dano adicional ao DNA.
Câncer que gera muito inflamação vai crescer mais rápido → Consequência de crescimento rápido é o aumento de radicais livres → Consequência de aumento de radicais livres é o aumento das mutações.
Gene APC → Gene supressor de tumor
Beta-catenina → Pode translocar e ativar a proliferação celular
WNT → Fator de crescimento solúvel
Via de sinalização WNT/B-catenina compõe a regulação de uma grande variedade de processos fisiológicos em diferentes espécies, com um papel decisivo no desenvolvimento embrionário, proliferação e diferenciação celular.
Evasão neoplásica → Resposta celular NK/CD8:
1- Mutações em B2-microglobulina (célula neoplasica pode realizar a mutação na região b2-microglobulina onde não consegue comunicar).
2- 2- Célula tumoral produz o ligante para PD1 através do mecanismo de autorregulação.
Normal: APC fagocita uma célula mutada e apresenta a partir do MHC de classe I. Liga PD1 ao ligante PD1 e libera grânulos citotóxicos levando a morte da célula afetada.
A via da WNT e B-Catenina (fator de transcrição, junto ao TCF) muito usada em câncer de cólon, mutação de APC
Anticorpos monoclonais podem bloquear essa proteína da célula tumoral impedindo que essa realize a evasão do sistema imune através da ligação PD1 e PD1L, ligando ao PDL1 primeiro.

*A expressão da telomerase está diretamente relacionada a gravidade do câncer pois é um mecanismo de evasão da catástrofe mitótica, levando ao aumento de mutações e de proliferação aumentada das células cancerígenas em questão.
Bibliografia: Anotações em aula, material do aluno.
Reflexão: Hoje achei que meu desempenho foi positivo. Realizei o estudo do material, mesmo tendo que ir dormir tarde, pois estava com muita demanda pessoal. Continuo achando o conteúdo um pouco denso, de forma que precisa sempre de uma atenção extra. A primeira parte da aula foi bem explicada, porém achei a segunda metade um pouco acelerada, devido ao tempo, o que gerou de certa forma uma confusão em alguns momentos. Esse estudo em casa te se tornado cada vez mais necessário, como a revisão dele quando chego em casa pela noite. Garante uma maior retenção de tudo, mas não minto que de certa forma estou um pouco preocupado com essa matéria.


Comentários